
MET and TP 53: How They Relate to ALK Lung Cancer
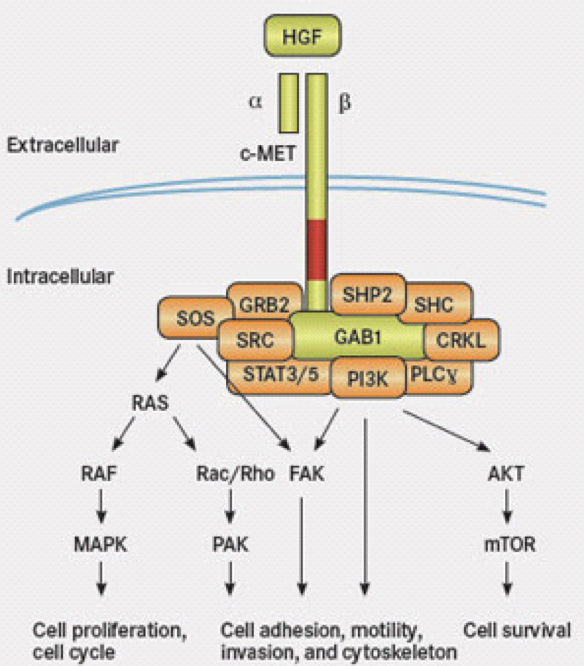
MET Amplification
MET is a receptor tyrosine kinase that participates in the hepatocyte growth factor (HGF)-mediated signaling pathway. This protein is anchored to the cell wall to pass information from the outside the cell to the inside of the cell. Normally, the receptor is activated by binding to HGF. This binding leads to phosphorylation (adding phosphate) of various sites on MET dimer (the complex of a and b MET) which in turn alters the complex to “open” up its binding domains for its downstream partners such as GAB1, GRB2 and so on.
Studies have been done in EGFR lung cancer patients showing resistance to a TKI (tyrosine kinase inhibitor, such as gefitinib for EGFR) can be due to MET amplification or MET hyperactivation. Extra MET leads to an increased activation of a downstream molecule called ErbB3. As the diagram above shows, there is a complex receptor system on the cell surface to receive information from outside the cell. There are many redundancies in the system to prevent potential problems from occurring naturally. However, this redundancy also makes it easier for cells to bypass specific inhibition of one of the receptors such as EGFR alone.
Researchers are looking to target specific proteins along the pathway to reduce tumor growth.

For MET amplifications, there are many known inhibitors such as crizotinib (or Xalkori, hooray!), tivantinib, savolitinib, tepotinib, cabozantinib, and foretinib. Not all are known in the “lung cancer” world. These have been used in other cancers to inhibit MET. There are ongoing clinical trials for combination treatment of EGFR-MET at this time. Perhaps this will extend to ALK-MET patients in the future.
Read more:
https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-018-0793-1
https://jhoonline.biomedcentral.com/track/pdf/10.1186/s13045-019-0759-9
Mutated Gene TP 53
This is a review done in 2017.
Published abstract:
TP53 is the most frequently mutated gene in human cancer. Functionally, p53 is activated by a host of stress stimuli and, in turn, governs an exquisitely complex anti-proliferative transcriptional program that touches upon a bewildering array of biological responses. Despite the many unveiled facets of the p53 network, a clear appreciation of how and in what contexts p53 exerts its diverse effects remains unclear. How can we interpret p53’s disparate activities and the consequences of its dysfunction to understand how cell type, mutation profile, and epigenetic cell state dictate outcomes, and how might we restore its tumor-suppressive activities in cancer?
Summary:
This was a protein of 53KD (Kilo-Dalton, a measurement for protein size) found in 1979. The normal p53 protein functions to regulate cells and the mutation in the TP53 gene (that encodes the p53 protein) leads to many cancers. Mouse model with no functioning p53 can developed normally but had high incidence of tumor.
This is a “picture” of p53.

The white glob is the p53 protein. The green and the purple is the DNA strands. So, p53 binds to DNA to open it up for transcription whenever it is needed. The red spots are the common places where p53 are often mutated or changed in tumor cells. Many tumors contain changes in the DNA binding domain of p53.
https://pdb101.rcsb.org/motm/31
This protein is highly regulated by many other cellular functions and only low amount is present in the cell at one time. The protein is degraded by ubiquitin ligase (adds ubiquitin) and is stabilized by phosphorylation by other kinases (enzymes that adds phosphate). There are various post-translational modifications to p53 for regulation. Cellular stress, DNA damage, replication stress can all lead to changes in p53 protein levels in the cells. So, what happens when p53 is induced? P53 can lead to cell cycle arrest or cell death. From the volumes of research, p53 is a very complex transcription factor that is used by many cell types for response to external factors. The nature of how p53 binds to DNA to regulate other downstream genes is very complicated with multiple variations of kinetics at the binding site, the post-translational modifications, and binding strength. It is not a simple “on-off” switch. This protein has also been implicated in cell fate determination. There are a family of proteins similar in nature to p53, called TP63 and TP73. In a triple null mouse model (no p53, p63, p73), meso-endodermal cell fate is disrupted. It appears p53 is integral in stem cell biology and pluripotency. Pluripotency means a cell that can become many different cell types. In the example of wound regeneration, p53 is important in extracellular matrix remodeling, proliferation of cells to regenerate, and resolution of fibrosis. Too little p53 can lead to early onset of cancer, and too much p53 can lead to early aging according to a mouse model.
Many researches since 2011 have been attempting to tackle the problem of controlling p53. One idea is to stop a molecule that controls p53 to stop p53 activation. Another method is to add a drug to re-gain the normal p53 function. Many of these studies are still on-going.
https://www.cell.com/cell/pdf/S0092-8674(17)30953-4.pdf